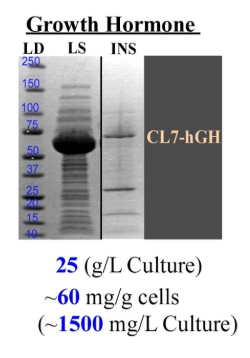
In 2013, Dmitry Vassylyev, a crystallographer at UAB, faced challenges in purifying proteins for crystallography due to the lack of a single affinity chromatography system providing ultra-high purity proteins. Existing methods suffered from flaws like sensitivity to high salt buffers, tag retention, low resin capacity, or high cost. To address this, Vassylyev devised a new approach, utilizing Colicin DNases and their inhibitors. By modifying the CE7 variant to retain binding affinity but lose toxicity, a novel CL7/Im7 system was created, where CL7 acts as a tag and Im7 as the immobilized ligand. This innovative system was detailed in a 2017 PNAS publication, offering a promising solution to the unmet need for efficient protein purification.

True Potential of Protein Purity for Structural Studies: A CLīM™ Technology Perspective
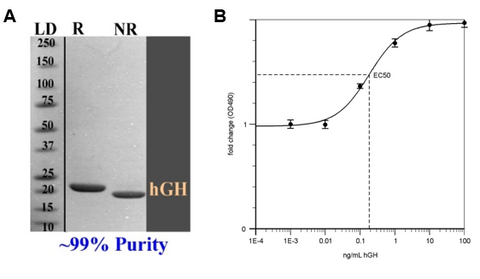
In the pursuit of protein purification, the ultimate goal is to attain a level of purity that aligns with the specific application at hand. The landscape of structural biology demands a meticulous approach, necessitating purity levels exceeding 95%. This shift is particularly pronounced in studies involving X-ray crystallography or cryo-EM, where a purity range of 95% to 99% is imperative for accurate structural insights.
Compromise on protein purity introduces inherent risks. In techniques such as X-ray crystallography, impurities hinder crystal formation, resulting in poorly diffracting crystals and low-resolution structures. Cryo-EM encounters challenges in processing images and suffers from reduced structure resolution with poorly purified proteins. Similarly, NMR spectroscopy struggles with impure proteins, making it challenging to distinguish target protein signals accurately.
The revolutionary CLīM™ technology emerges as a streamlined solution for structural biologists. CLīM™ ensures unparalleled protein purity through a one-step chromatography process, eliminating the need for extensive multistep protocols. With its high binding capacity Im7 resin, CLīM™ technology results in efficient purification, rivaling the traditional approach of using multiple columns for different purification steps.
To achieve the pinnacle of purity for structural biology applications, TriAltus recommends the CLīM™ system over conventional chromatography methods.
TriAltus Custom Protein Purification Service for Structural Biologists:
Our custom protein purification service offers structural biologists the opportunity to delegate key projects, ensuring the highest purity and integrity of proteins for advanced structural studies. Additionally, upon request, we are fully equipped to deliver proteins in compliance with ISO 13485 standards, meeting the highest quality and regulatory requirements. Partner with TriAltus to accelerate your research while maintaining uncompromised excellence.
Contact us for a personalized consultation.
Email: info@trialtusbioscience.com
Phone: +1.205.453.8242
]]>